LACE: Inference of cancer evolution models from longitudinal single-cell sequencing data
Daniele Ramazzotti, Fabrizio Angaroni, Davide Maspero, Gianluca Ascolani, Isabella Castiglioni, Rocco Piazza, Marco Antoniotti, Alex Graudenzi
Journal of Computational Science, February 2022

Abstract. The rise of longitudinal single-cell sequencing experiments on patient-derived cell cultures, xenografts and organoids is opening new opportunities to track cancer evolution, assess the efficacy of therapies and identify resistant subclones.
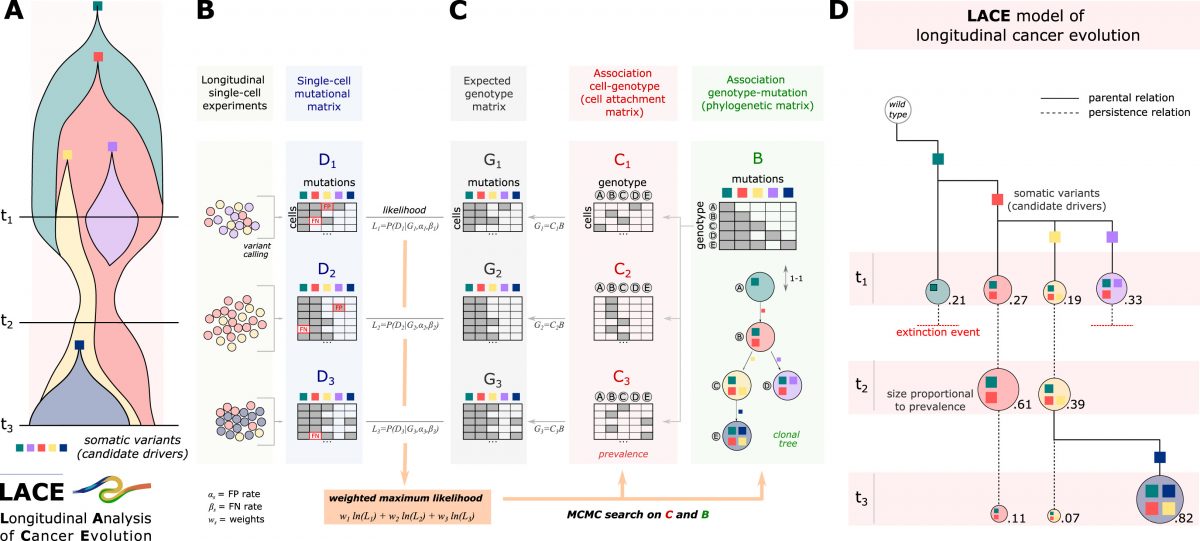
We introduce LACE, the first algorithmic framework that processes single-cell mutational profiles from samples collected at different time points to reconstruct longitudinal models of cancer evolution. The approach maximizes a weighted likelihood function computed on longitudinal data points to solve a Boolean matrix factorization problem, via Markov chain Monte Carlo sampling.
On simulations, LACE outperforms state-of-the-art methods for both bulk and single-cell sequencing data with respect to the reconstruction of the ground-truth clonal phylogeny and dynamics, also in conditions of unbalanced datasets, significant rates of sequencing errors and sampling limitations. As the results are robust with respect to data-specific errors, LACE is effective with mutational profiles generated by calling variants from (full-length) scRNA-seq data, and this allows one to investigate the relation between genomic and phenotypic evolution of tumors at the single-cell level.
Here, we apply LACE to a longitudinal scRNA-seq dataset of patient-derived xenografts of BRAFV600E/K mutant melanomas, dissecting the impact of BRAF/MEK-inhibition on clonal evolution, also in terms of clone-specific gene expression dynamics. Furthermore, the analysis of breast cancer PDXs from longitudinal targeted scDNA-sequencing experiments delivers a high-resolution temporal characterization of intra-tumor heterogeneity.