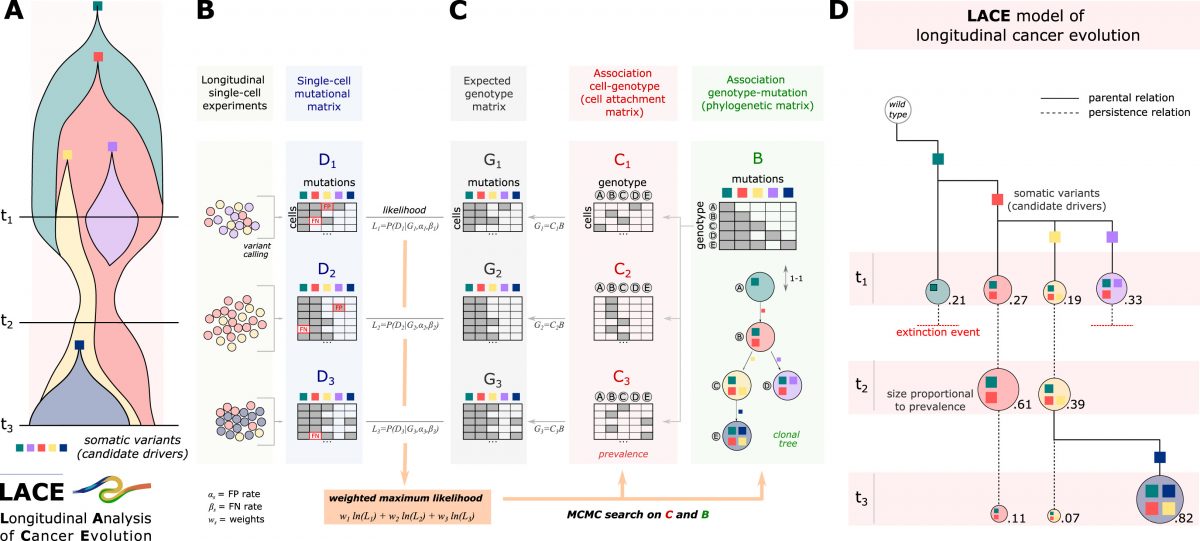
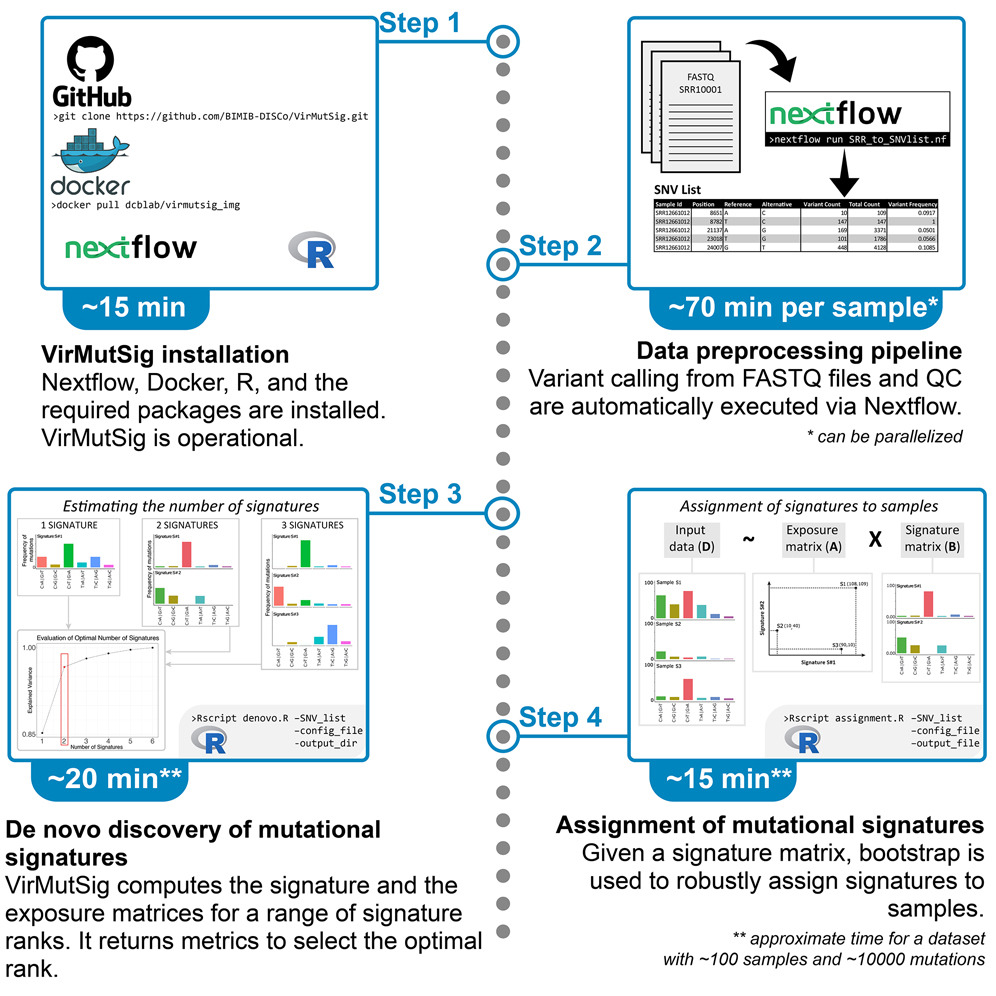
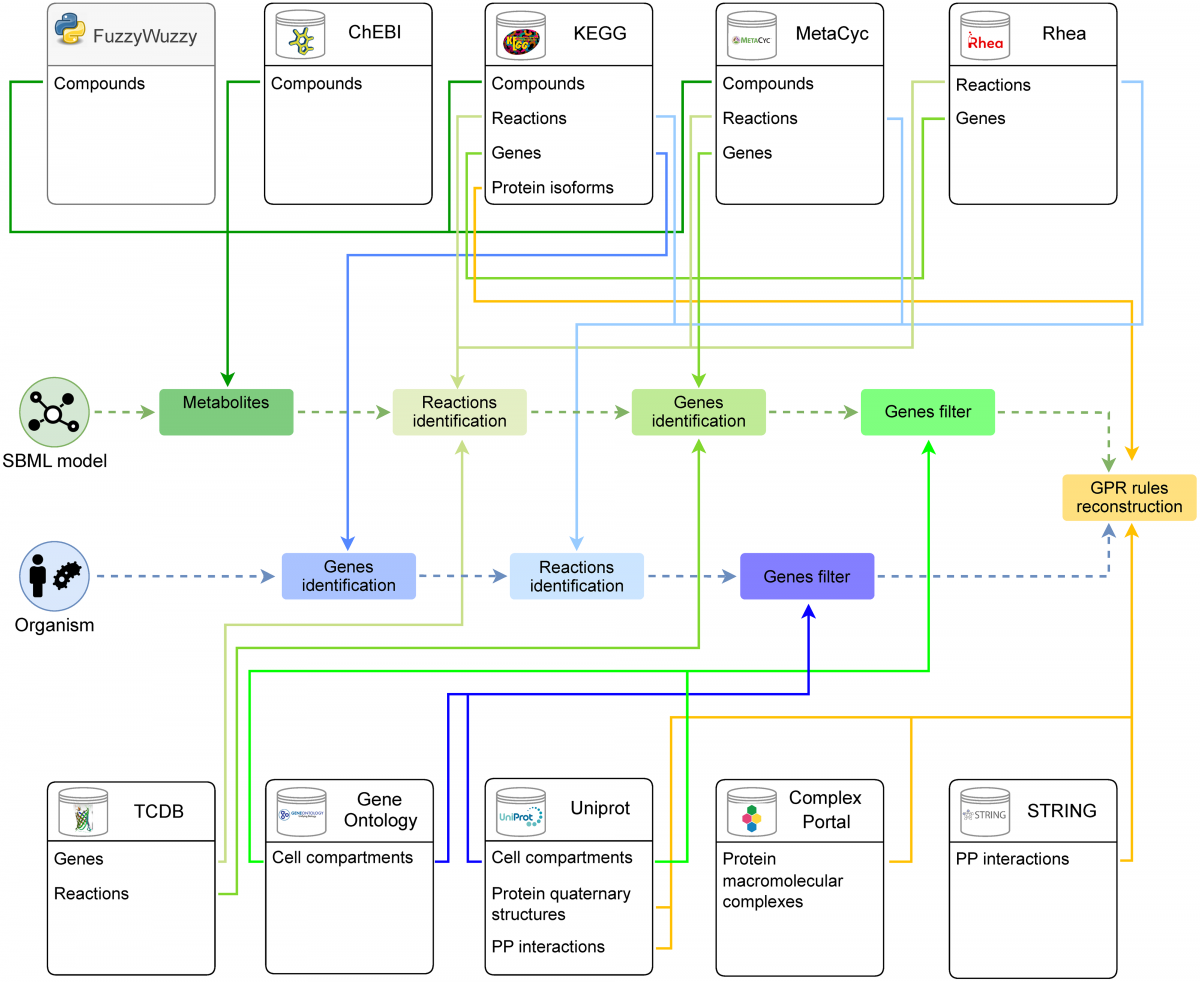
CSCE 2025
Cancer is a multi-factorial disease caused by the malfunction of the bio-molecular machinery that regulates the body’s “checks and balances”. This leads to the uncontrolled growth of certain cell subpopulations selected by evolutionary pressure, which ultimately threatens the host’s survivaI.
In the last 15 years, countless algorithmic, statistical, and mathematical modelling strategies have greatly aided in understanding the disease’s intricacies, especially by leveraging the vast and increasing amounts of omics data generated from cancer samples. Importantly, new experimental paradigms, such as those on patient-derived models are delivering the first exciting results.
In this lively field, the Como School on Cancer Evolution (CSCE 2025) brings together researchers from both dry- and wet-labs to explore the challenges posed by cancer as a an evolutionary disease.
The School will allow the participants to gain expertise on state-of-the-art concepts, methods and applications from both cancer biology and computational sciences, especially data science and artificial intelligence, and to get a glimpse into the vision of pioneers in the field of cancer evolution.