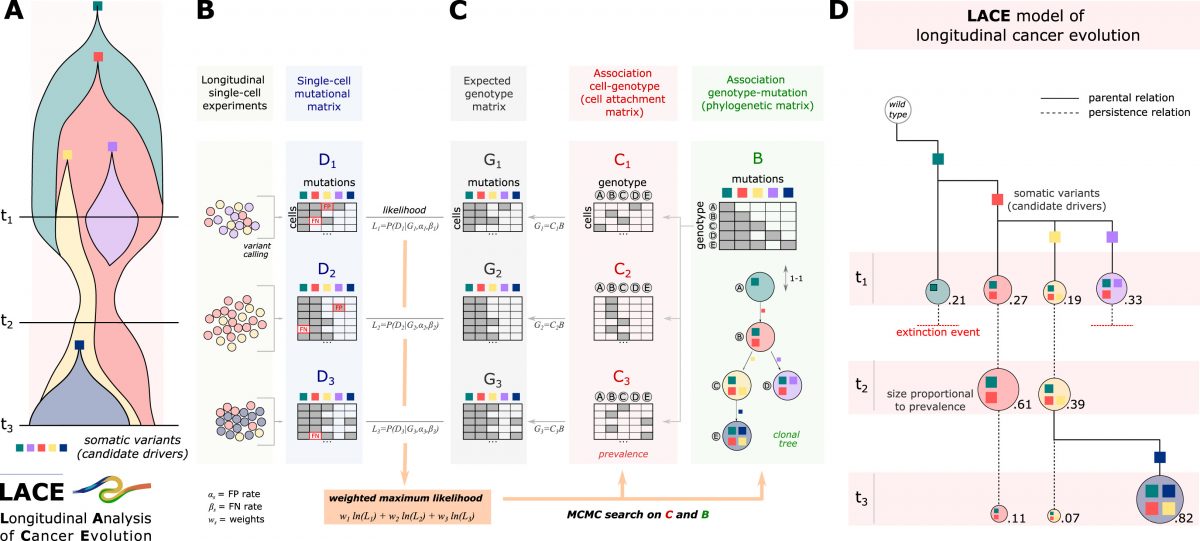
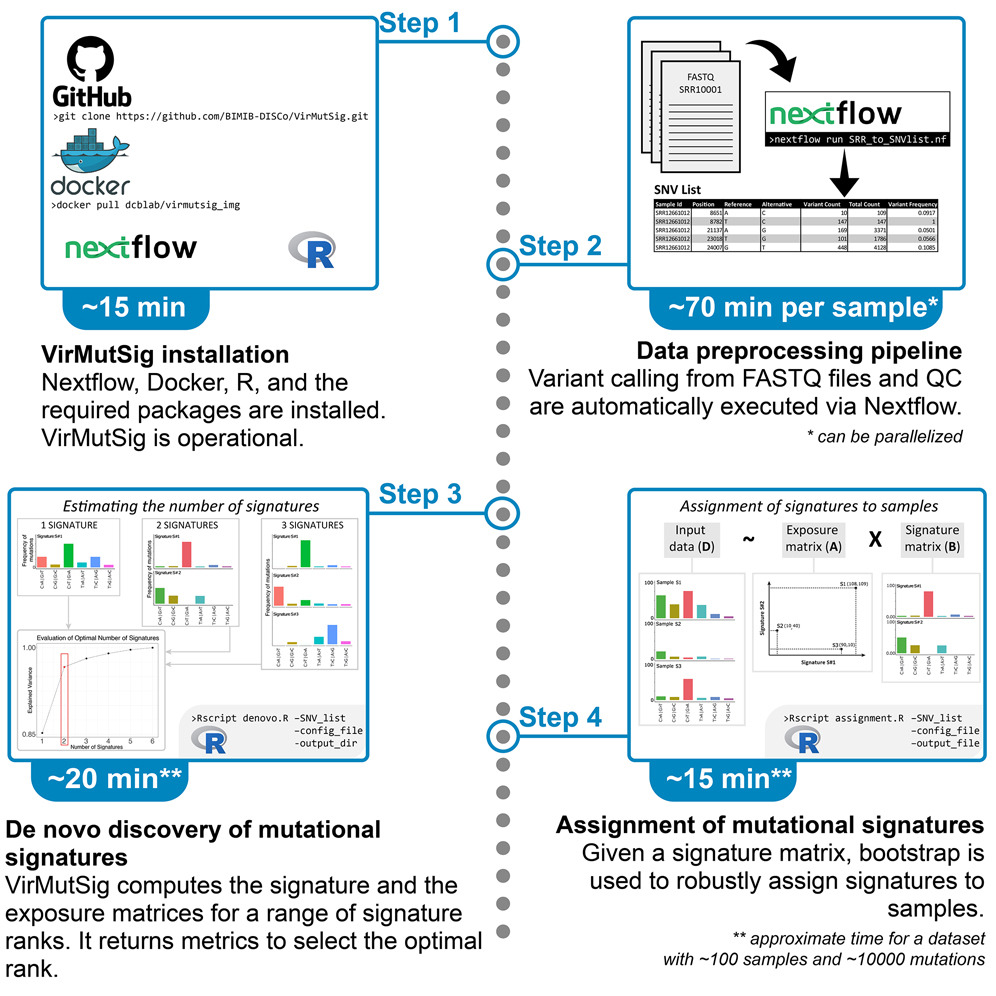
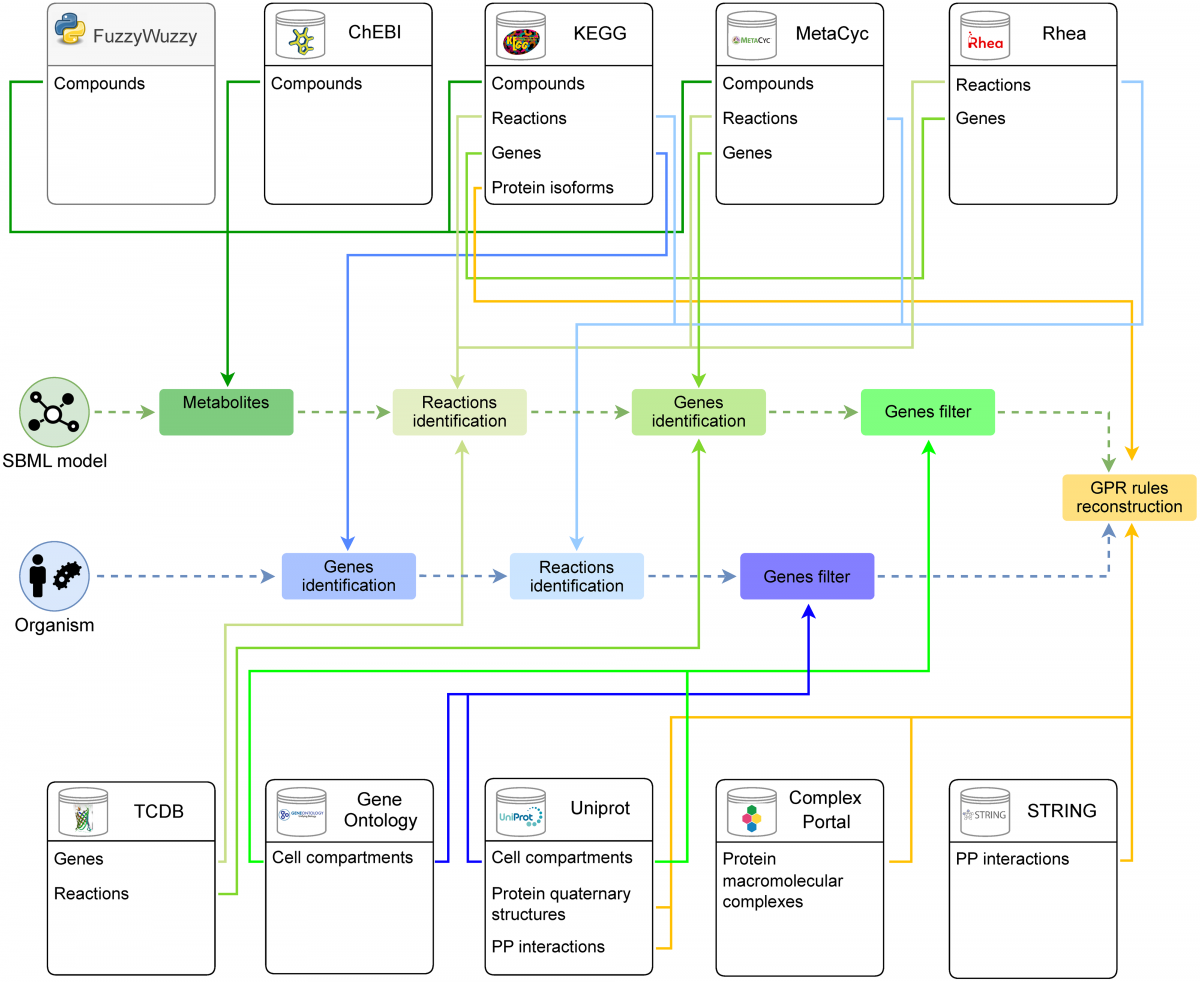
Large-scale analysis of SARS-CoV-2 synonymous mutations reveals the adaptation to the human codon usage during the virus evolution
Daniele Ramazzotti, Fabrizio Angaroni, Davide Maspero, Mario Mauri, Deborah D’Aliberti, Diletta Fontana, Marco Antoniotti, Elena Maria Elli, Alex Graudenzi, Rocco Piazza
Virus Evolution, Volume 8, Issue 1, 2022
DOI: https://doi.org/10.1093/ve/veac026
⚠️ Interview by Luca Salvi, BNews, to Daniele Ramazzotti (link).
Abstract. Many large national and transnational studies have been dedicated to the analysis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) genome, most of which focused on missense and nonsense mutations. However, approximately 30 per cent of the SARS-CoV-2 variants are synonymous, therefore changing the target codon without affecting the corresponding protein sequence.
By performing a large-scale analysis of sequencing data generated from almost 400,000 SARS-CoV-2 samples, we show that silent mutations increasing the similarity of viral codons to the human ones tend to fixate in the viral genome overtime. This indicates that SARS-CoV-2 codon usage is adapting to the human host, likely improving its effectiveness in using the human aminoacyl-tRNA set through the accumulation of deceitfully neutral silent mutations.
One-Sentence Summary. Synonymous SARS-CoV-2 mutations related to the activity of different mutational processes may positively impact viral evolution by increasing its adaptation to the human codon usage.