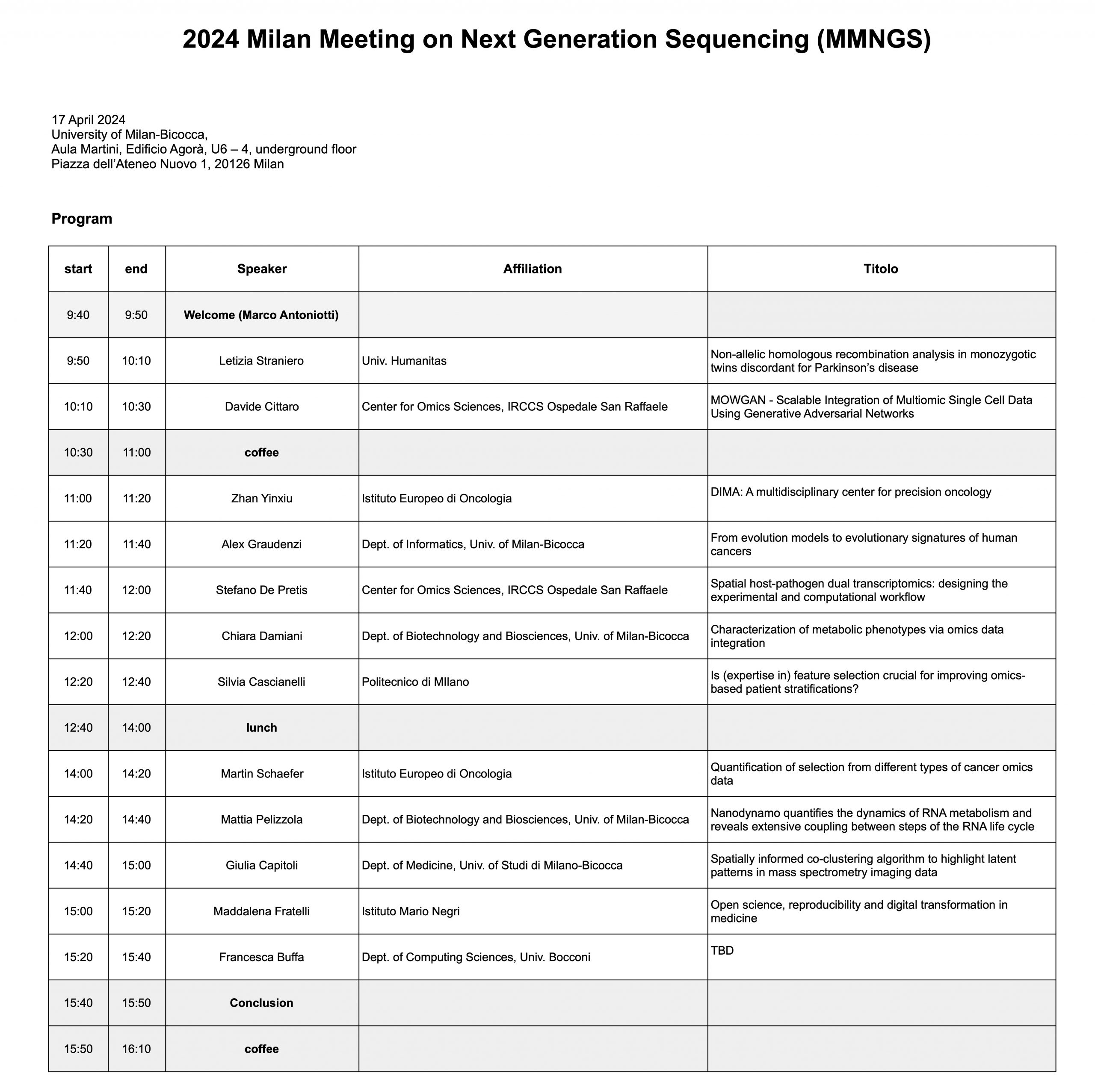
23 Maggio 2024, ore 10:30 – 12:00
Seminar room, Abacus Building (U14)
Department of Informatics, Systems and Communication
Speaker:
Prof. Iuliana Ionita-Laza
Department of Biostatistics, Columbia University, New USA
and Department of Statistics, Lund University, Sweden
Abstract:
Many complex traits such as schizophrenia, Body Mass Index and various cancers have a substantial heritable component as evidenced by the large number of genetic risk variants discovered by genome-wide association studies (GWAS). Despite these successes, a substantial gap remains between our current understanding and the ability to translate this knowledge into targeted treatment strategies.
A major limitation to the conventional GWAS approaches is the reliance on oversimplified statistical models that fail to capture the complexity underlying genotype-phenotype associations. For example, many human diseases are age-related, and many genetic associations are also age dependent. Such underlying complexity induces heterogeneity in genetic associations and linear regression models in GWAS are not well suited to decipher such heterogeneous associations.
In this talk I will discuss alternative methods based on quantile regression for GWAS and polygenic risk score (PRS) prediction, and will show applications to diverse traits in the UK biobank.
Contact person for this Seminar: marco.antoniotti [at] unimib.it